4/ We implemented MCS in the read aligner strobealign.
Result:
- Higher mapping accuracy
- Little to no runtime overhead
- No additional memory
09.03.2026 12:22
👍 0
🔁 0
💬 0
📌 0

@ksahlin
Associate Professor at the Department of Mathematics, Stockholm University, and a Scilifelab Fellow. Algorithms, Modeling, Transcriptomics, Genomics. Hobby runner 5000m 18:48 | 10k 37:40 | HM 1:27:43 | M 3:39:06
4/ We implemented MCS in the read aligner strobealign.
Result:
- Higher mapping accuracy
- Little to no runtime overhead
- No additional memory
3/ We introduce Multi-Context Seeds (MCS).
Idea: represent seeds at multiple resolutions by partitioning the bits of the hash value. Identical smaller seeds share prefix. Using a sorted (by hash value) flat vector index with a prefix-lookup vector*, switching resolutions is fast.
2/ In sequence mapping, there's a classic tradeoff:
Long seeds → fast but less sensitive
Short seeds → sensitive but slower
How can we get both speed and sensitivity?

1/ Our paper on Multi-Context Seeds is now out, with @tolyan.bsky.social spearheading the work and contributions from Nicolas and @marcelm.net. We introduce a new seeding concept that improves read alignment accuracy while maintaining speed.
link.springer.com/article/10.1...

Excited to share this preprint that describes my latest work on using GPUs to accelerate processing of RNA-seq data.
The title says it all: "RNA-seq analysis in seconds using GPUs" now on biorxiv www.biorxiv.org/content/10.6... and github github.com/pachterlab/k...
Figure 1 shows they key result
DSB Program is out !
Seems incredible (as ever)
dsb-meeting.github.io/DSB2026/

... and the preprint has also been updated: www.biorxiv.org/content/10.1...
The commit benefited tremendously by minimap2’s practical algorithmic solutions for chaining and rescue -- big credits to @lh3lh3.bsky.social and his minimap2. Also to my students Nicolas Buchin and Ivan Tolstoganov, as well as Marcel Martin 4/4
Strobealign can now map long reads. Still POC, i.e., PAF only -- no supplementary chains or piecewise extension (yet). 3/4
Accuracy upgrades: chaining instead of NAMs, smarter local rescue of repetitive hits (minimap2-style), and improved multi-context seeding 2/4
Thank you folks for your feedback on our survey about Hash functions in genomic sequence analysis. We've updated the paper and you can see the new version here: tinyurl.com/4kk9ccmt.
Preprint out for myloasm, our new nanopore / HiFi metagenome assembler!
Nanopore's getting accurate, but
1. Can this lead to better metagenome assemblies?
2. How, algorithmically, to leverage them?
with co-author Max Marin @mgmarin.bsky.social, supervised by Heng Li @lh3lh3.bsky.social
1 / N


Congratulations to Rayan Chiki, (Institut Pasteur) head of the “Sequence Bioinformatics” unit, for securing the ERC Proof of Concept 2025 for his project ENZYMINER! 👏
@rayan.chiki.bsky.social
#Bioinformatics
Incredible! 👏


We have officially started #HitSeq track @hitseq.bsky.social at #ISMBECCB2025. Francisco de la Vega, introduces our first #keynote speaker Valentina Boeva @valboeva.bsky.social with her talk: "Learning variant effects on chromatin accessibility and 3D structure without matched Hi-C data"
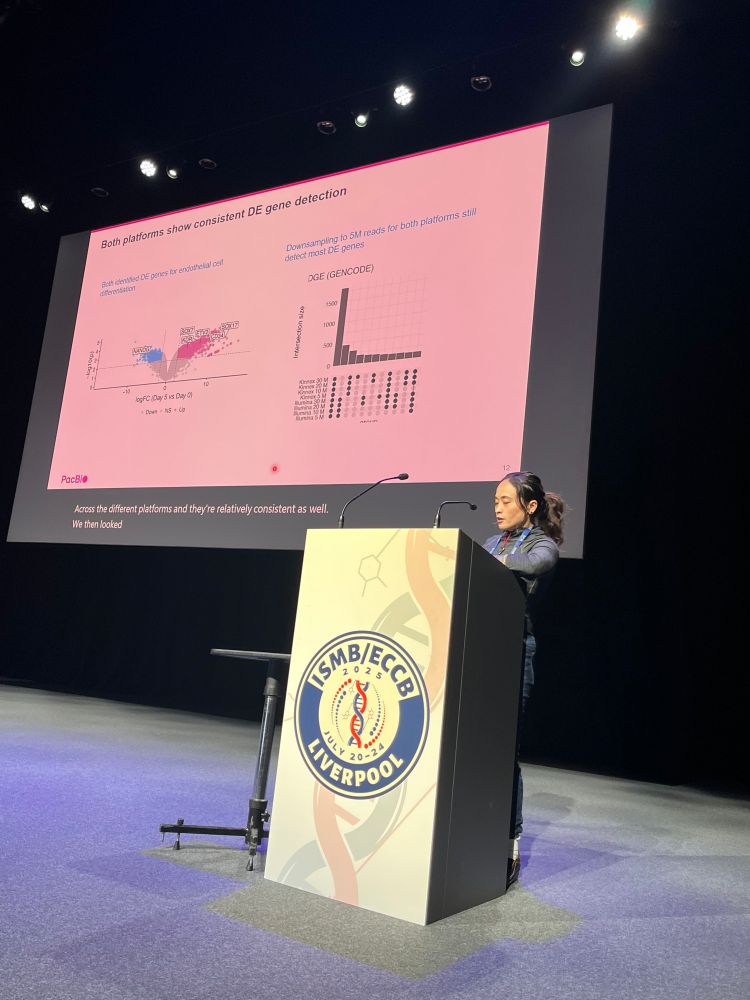
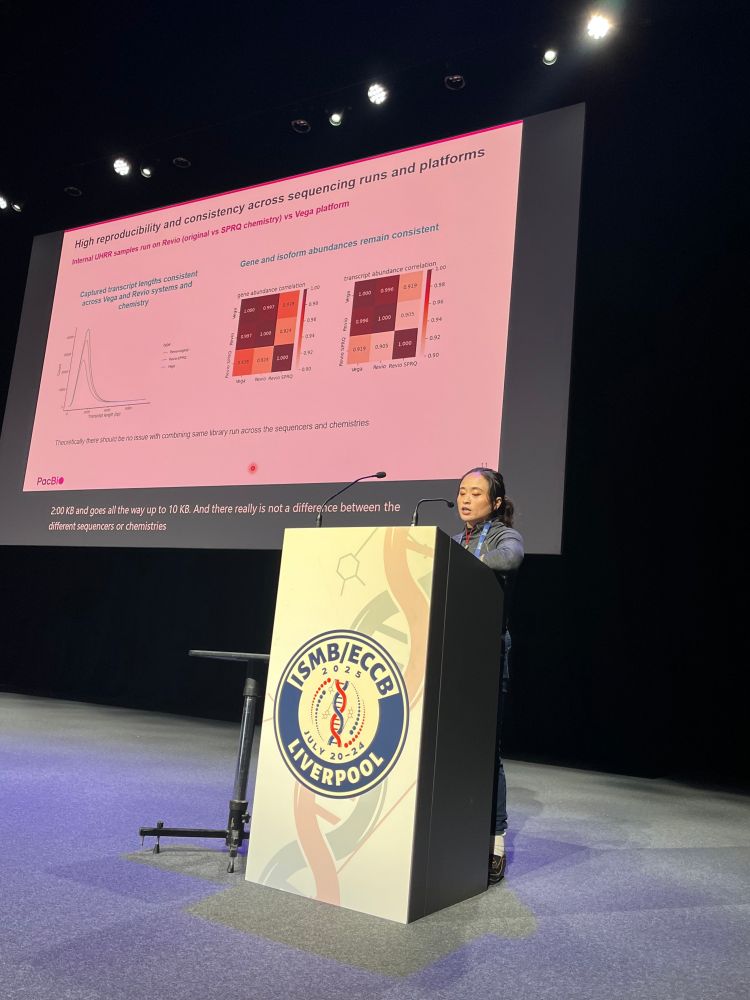
Meet our amazing sponsor PacBio @pacbio.bsky.social for @hitseq.bsky.social track at #ISMBECCB2025 represented by Elizabeth Tseng with her talk "Bioinformatics analysis for long-read RNA sequencing: challenges and promises" #hitseq #iscb #sequencing #application #iverpool #uk

Dont miss any of our #LongTREC communications at #ISMBECCB2025. Download this flyer to make catching all the latest & hottest long-read transcriptomics research simple.
@anaconesa.bsky.social


@hitseq.bsky.social is kicking off with our first keynote @valboeva.bsky.social talking about "Learning variant effects on chromatin accessibility and 3D structure without matched Hi-C data". #ISMBECCB2025
📽️ Next in the LongTREC Series: Mahmud Sami Aydin!
Sami is a Doctoral Candidate at @stockholm-uni.bsky.social , working under the supervision of @ksahlin.bsky.social .In this video, Sami shares his research and his role in the broader LongTREC collaboration across Europe.
#AlgorithmDevelopment

Paper alert!
We present Oreo a tools that reorder long reads datasets in a way to compress them efficiently with ANY universal compressor like gz, zstd, xz ...
TLDR: You can get state of the art compression WITHOUT a dedicated compressor/decompressor!
academic.oup.com/bioinformati...
A thread!
I worked with Thomas during a three months research visit during his PhD, and it resulted in a paper in NAR. I highly recommend him. doi.org/10.1093/nar/...

Thomas Baudeau defended his thesis on Studying the properties of viral long reads mapping methods - congrats docteur Baudeau you'll be deeply missed in the team. I'm very glad I got the chance to work with you. Thomas is also on the lookout for a postdoc 👀
🧵1/n
Estimating mutation rates using k-mers is fast—but what happens when repeats dominate the genome?
In a new preprint, Haonan Wu, Antonio Blanca, and myself propose a *repeat-aware* estimator that's accurate even in centromeres.

Hey yeast lovers. Do you like pangenomes?
O'Donnel et al. 2023 produced T2T assemblies of different strains, including phased haplotypes for yeast.
Here I selected 10 phased haplotypes and the S288C reference,
and looked for the MST28 / YAR033W gene reported to contain SVs such as indels.
👇🏻👇🏻
Congrats 👏👏
IMO it matters a lot as a 'first impression'
I did only very minor impl. contributions, but from my (non-expert) view, I like that (1) it installs easily (also on a MacBook) and (2) no header files. Felt much easier to get started with than, e.g., C++. I never truly learned good .h/.cpp practices, and I could never get OpenMP/g++ working well

