Since Yb is Drosophilid-specific, we don't expect UU-sensing to be conserved directly. In mammals, specificity is primarily driven by ping-pong.
There are other piRNA independent systems that depend heavily on the nucleotide composition (e.g. the HUSH complex targetting A-rich intronless RNAs)
16.02.2026 09:50
👍 0
🔁 0
💬 0
📌 0

13.02.2026 15:14
👍 8
🔁 0
💬 1
📌 0
Lastly, huge thanks to Julius (@juliusbrennecke.bsky.social, @rippei.bsky.social), Wylan (@chaoticmetazoan.bsky.social) , Aleks and the entire Brennecke Group (current and former members)!
This project has been a long journey, and we’re thrilled to finally share it with the community. 🧬🚀 (18/18🏁)
13.02.2026 15:11
👍 7
🔁 1
💬 2
📌 0
Excitingly, this isn’t limited to flies! 🌍
A preprint from the Tomari lab finds naïve piRNA biogenesis across various insects and mammals. It seems “sample everything, filter for the enemy” is a conserved strategy for genome defense against transposons. (17/18)
www.biorxiv.org/content/10.1...
13.02.2026 15:11
👍 5
🔁 3
💬 2
📌 0

Two-panel summary of germline ping‑pong depletion. Left: stacked bar chart of piRNA annotation categories comparing control versus Aub+Ago3 knockdown; TE-derived piRNAs drop and genic (CDS/3′UTR) fraction increases. Right: violin plot of Piwi-bound piRNA changes (Aub+Ago3 depletion / control) showing clusters decrease strongly while genic piRNAs increase or are less reduced, revealing a “hidden” basal genic piRNA population when ping‑pong is blocked.
Germline cells don’t express Yb; they use "Ping-Pong." We "unmasked" the basal machinery by blocking Ping-Pong (Aub/Ago3 depletion). The result: transposon piRNAs collapsed, and a "hidden" genic pool emerged. Naïve biogenesis is a shared foundation of both soma and germline! (17/19)
13.02.2026 15:11
👍 4
🔁 0
💬 1
📌 0
This reveals a naïve sampling core: a basal biogenesis program that is inherently non-selective and abundance-driven.
Specificity isn’t built into the core. Instead it’s layered on top by modules like Yb. But is this core universal? Does it also operate in the ping-pong driven germline? (16/19)
13.02.2026 15:11
👍 6
🔁 0
💬 1
📌 0

Model trained on Yb-depleted data. Left: scatter plot of observed vs predicted piRNA levels for 3′UTR tiles (log–log) showing good prediction. Right: feature-importance bars showing transcript abundance/position features dominate, while nucleotide-content features (A/C/G/T) contribute little—consistent with loss of sequence-based specificity without Yb.
To test this, we trained a new gradient boosting model specifically on this Yb-depleted data. The result? The nucleotide-content completely lost its predictive power. Instead, transcript abundance became the primary driver. The machinery simply samples what is most available. (15/19)
13.02.2026 15:11
👍 5
🔁 0
💬 1
📌 0

Small-RNA overview comparing control vs Yb-depleted cells. Left panels: size/abundance distributions showing piRNAs (23–35 nt) remain but change in amount. Right: stacked bar chart of piRNA annotations (CDS, 3′UTR, TE sense/antisense, etc.) showing a compositional shift in Yb depletion from TE-dominated piRNAs toward a genic (mRNA feature)–dominated pool.
What happens without the specificity factor Yb? The pathway doesn't collapse, but it now shifts from transposons to a genic-dominant piRNA distribution, revealing that the machinery’s default state is to process almost any available RNA indiscriminately. (14/19)
13.02.2026 15:11
👍 4
🔁 0
💬 1
📌 0

Genome-browser–style multi-track plot across a flamenco region with an orientation switch (antisense U-rich region into a sense, U-poor insertion). Tracks show transcription direction, local U-content, piRNA levels in control (high in U-rich/antisense region) versus Yb knockdown (reduced), and a log2 fold-change track that is strongly negative over the U-rich region. Overall message: sense (A-rich → U-poor in RNA) insertions are poor piRNA substrates; Yb is required for the strong U-dependent output.
Our model predicts this logic should work both ways: if a transposon is inserted in the sense orientation (A-rich, not U-rich), it shouldn't produce piRNAs efficiently, even in flamenco.
Indeed, a sense TE insertion in flamenco is a poor substrate. It's about the sequence, not the location. (13/19)
13.02.2026 15:11
👍 7
🔁 1
💬 1
📌 0
Yb senses this bias to find targets without a predefined template, even for "de novo" insertions in 3’ UTRs. 🧬
This allows the pathway to catch new threats anywhere in the genome, a mechanism beautifully demonstrated in my colleague Baptiste’s latest preprint! (12/19) bsky.app/profile/bapt...
13.02.2026 15:11
👍 4
🔁 1
💬 1
📌 0

Two stacked line plots comparing nucleotide composition along sequences (x-axis: scaled position; y-axis: nucleotide content %). Top: host 3′UTRs are AT/U-rich relative to GC. Bottom: LTR retrotransposon coding sequence composition shows strong A/T bias compared to GC, illustrating transposons are adenosine-rich at the DNA level (which becomes uridine-rich when transcribed antisense).
But why? How does a simple U-code target transposons? It’s a brilliant exploit of retrotransposon evolution: their genomes are intrinsically A-rich. When transcribed in antisense (as in clusters), they become exceptionally U-rich, placing them squarely in Yb’s crosshairs. 🎯 (11/19)
13.02.2026 15:11
👍 8
🔁 3
💬 1
📌 1

Two-panel figure for the flamenco piRNA cluster. Left: scatter plot comparing observed vs predicted (recalibrated values predicted using the UTR trained model) piRNA levels for cluster tiles on log scales, showing strong correlation along a diagonal. Right: line plot of piRNA abundance across flamenco position (kb) comparing observed and predicted profiles, with the two curves closely tracking across the locus.
The best part? Our model, trained only on 3'UTRs, accurately captured the piRNA profile of the flamenco cluster.
This proves flamenco isn't "special." It follows the same rules as regular genes—its massive piRNA biogenesis efficiency is simply a direct consequence of its extreme U-richness. (10/19)
13.02.2026 15:11
👍 5
🔁 3
💬 1
📌 0

Left: scatter plot of observed vs model-predicted piRNA levels for 3′UTR tiles (log–log axes) with points near a diagonal “perfect prediction” line, indicating strong predictive performance. Right: horizontal bar chart of model feature importance showing UU dinucleotide content and transcript abundance (TT-seq/RNA-seq features) as top predictors; other nucleotide-composition features contribute less.
To test if these features are sufficient to explain piRNA biogenesis, we trained a gradient boosting model on regular host 3'UTRs. The result? We achieved quantitative prediction of piRNA output, with UU-content and transcript abundance emerging as the dominant predictors. 🤖📊 (9/19)
13.02.2026 15:11
👍 6
🔁 0
💬 1
📌 0

Two piRNA profiles from reporters with the same overall U content (~33%) but different U arrangement. One configuration yields very low piRNAs across the reporter, while the other produces much higher piRNA levels (peaking around the central UTR region), illustrating that U patterning—especially UU dinucleotides—drives Yb-dependent piRNA output beyond total U percentage.
Is Yb just sensing simply uridine levels? Not quite. Using sensors with identical U-content but different configurations, we found that UU dinucleotides are key. It’s not just about the total count but more about the specific arrangement of uridines. (8/19)
13.02.2026 15:11
👍 5
🔁 0
💬 1
📌 0

“U-ramp” synthetic reporter assay. Left: bar/track-style piRNA profiles along a reporter with 20% U in the 3′UTR (near-zero piRNAs) versus 35% U (strong piRNA production peaking across the UTR). Right: scatter plot of total reporter piRNA counts versus 3′UTR uridine percentage showing a sharp threshold-like increase above ~30% U in control cells; Yb knockdown flattens/reduces the high-U response.
Check this out: we designed "U-ramp" reporters with entirely synthetic 3’ UTRs by varying in their uridine content (20% to 45%). The result was striking: U-density behaves like a threshold—below ~30% U, barely any piRNAs; above it, output shoots up in a Yb-dependent manner. (7/19)
13.02.2026 15:11
👍 17
🔁 4
💬 1
📌 0

Two-panel figure linking piRNA production to uridine. Left: distributions of piRNA biogenesis efficiency for CDS and 3′UTR tiles grouped into efficiency bins (log scale). Right: violin plots showing uridine content (%) in the analysis window for each efficiency bin; higher-efficiency bins have higher U content, indicating U-rich sequence associates with stronger piRNA output.
We discovered that Yb acts as a U-content sensor. It selectively channels uridine-rich transcripts to piRNA biogenesis, effectively initiating their phased processing into piRNAs. (6/19)
13.02.2026 15:11
👍 7
🔁 4
💬 1
📌 0

Two-panel figure. Left: stacked bars comparing from which across feature classes (CDS, 5′UTR, 3′UTR, unannotated, TE sense, TE antisense) piRNAs or RNAseq reads originate; piRNAs are enriched for TE antisense and 3′UTR relative to RNA-seq. Right: violin/dot plots of “piRNA biogenesis efficiency” (piRNA output normalized to RNA abundance or transcription) showing cluster regions are far more efficient than typical 3′UTRs, and CDS regions are least efficient; y-axis is log-scaled.
In ovarian somatic cells (OSCs), piRNAs don't just come from the famous flamenco cluster: ~20% map to thousands of host mRNA 3′UTRs/CDSs (first shown by Lai & Lau).
We used this genic pool to decode Yb - what sequence features determine how efficiently a transcript becomes a piRNA source? (5/19)
13.02.2026 15:11
👍 6
🔁 0
💬 1
📌 1
The answer lies in the DEAD-box protein Yb.
The Siomi labs pioneered our understanding of Yb as a potential specificity factor—but what molecular signal does it actually read out? We set out to decode the logic Yb uses to "license" transcripts for processing. 🔎 (4/19)
13.02.2026 15:11
👍 4
🔁 0
💬 1
📌 0
We started by analyzing a piRNA pathway that lacks the well-known "Ping-Pong" amplification cycle that steers piRNA biogenesis in the germline. Somatic cells in the Drosophila ovary lack ping-pong, leaving a major black box: how does this pathway achieve specificity against transposons? (3/19)
13.02.2026 15:11
👍 4
🔁 0
💬 1
📌 0
Schematic of piRNA biogenesis in a Drosophila ovariole highlighting a shared “naïve RNA sampling” core in soma and germline. In both tissues, cytoplasmic transcripts can enter Piwi/Zucchini-dependent phased piRNA production randomly. Specificity is added by tissue-specific modules: in soma a slicer-independent module centered on Yb recognizing U-rich RNAs/TE antisense; in germline a slicer-dependent “ping‑pong” amplification cycle (Aub/Ago3) between TE sense and antisense transcripts that feeds into phased biogenesis.
We discovered that specificity of the pathway is not "hard-coded" in clusters. Instead, a naïve RNA sampling core in cells makes piRNAs from literally every cytoplasmic RNA. 🧬
Specificity then emerges from molecular modules that layer on top of the naïve core - but how does this work? (2/19)
13.02.2026 15:11
👍 10
🔁 2
💬 1
📌 0
How does the piRNA pathway solve the self vs. non-self problem? 🧬
Since piRNAs come from single-stranded RNA, how does the cell choose the right ones? For years, "piRNA clusters" were seen as THE privileged source. But are they really special and earmarked for biogenesis? (1/19)
13.02.2026 15:11
👍 90
🔁 51
💬 2
📌 4
🪱 Selfish genes are everywhere and drive some of biology’s biggest innovations (CRISPR, antibody recombination, epigenetics). Yet almost no one asks the obvious question: how does a selfish gene begin? Our new manuscript uncovers how selfishness can emerge directly from the host genome.
24.11.2025 13:03
👍 61
🔁 38
💬 1
📌 1
Intrigued by a long-standing conundrum in small RNA biology—how nuclear Argonaute proteins silence transposons when they *need* target transcription for their own recruitment—we studied the piRNA pathway.
And found a hidden RNA-decay axis from Piwi to the RNA exosome.
22.12.2025 18:14
👍 98
🔁 42
💬 3
📌 5

How does messenger RNA (mRNA) get out of the nucleus to become a protein? Eukaryotic mRNA is packaged, exported, and then translated in the cytoplasm. But how do these steps work? And what are open questions? Check out our new review for our take: www.annualreviews.org/content/jour... (1/3)
21.11.2025 17:36
👍 122
🔁 52
💬 1
📌 3
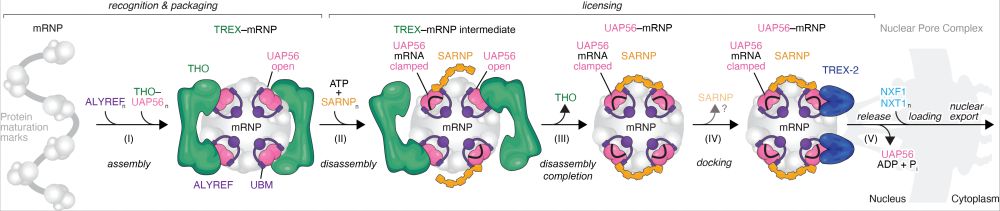
Finally out in @nature.com! We uncovered a mechanistic framework for a general and conserved mRNA nuclear export pathway. www.nature.com/articles/s41.... 1/
19.11.2025 23:21
👍 106
🔁 42
💬 3
📌 1
Are you an early-stage graduate student (2nd or 3rd year) or early-stage postdoc based in the US or Canada, working primarily in Drosophila? Would you like to help improve the experience of all trainees working in Drosophila research? If so, read on.
(Please repost to reach a broad audience.)
12.11.2025 04:49
👍 89
🔁 182
💬 1
📌 2
just in time for the opening of the @hohmannulrich.bsky.social group at @imbmainz.bsky.social
what started as a project on how cells export piRNA precursors, ended up as a tour de force in mRNA export. truly wonderful collaboration with @plaschkalab.bsky.social at the @viennabiocenter.bsky.social
07.11.2025 18:03
👍 32
🔁 11
💬 0
📌 0
No problem at all! I'm happy to clarify any questions that arise.
15.10.2025 10:50
👍 1
🔁 0
💬 0
📌 0
We used the term in this thread to distinguish this assembly from contig-only assemblies. If the usage of 'chromosome-scale' was misleading, we apologize, there was no intent to mislead.
15.10.2025 10:37
👍 1
🔁 1
💬 1
📌 0
We referred to it as chromosome scale as it is assembled into chromosome-level scaffolds spanning entire chromosome arms. We clearly state in the text that it is not telomere-to-telomere (T2T) and does not fully resolve highly repetitive loci like rDNA arrays or centromeres.
15.10.2025 10:37
👍 1
🔁 1
💬 1
📌 0
